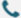General Introduction of Protein Structure Analysis
Structural biology services focus on studying the tertiary structure of biomacromolecules through physical methods, predicting and analyzing their functions, and uncovering the molecular mechanisms of macromolecular interactions.
At Synbio Technologies, we offer three advanced structural analysis platforms: MicroED, CryoEM-SPA (CryoEM single-particle analysis), and X-ray diffraction (XRD) for single-crystal analysis. These platforms provide tailored solutions based on the unique properties of different proteins. With state-of-the-art experimental equipment and a highly skilled, experienced research team, we deliver comprehensive structural analysis services starting from protein expression, designed to meet the diverse needs of both scientific research and industrial applications.
Highlights
-
![]()
- 750+ Projects Completed
Serving 350+ clients with 750+ successful project solutions.
-
![]()
- Fast Delivery
With weekly synchrotron radiation access and streamlined processes, projects are delivered in just 1 to 2 months.
-
![]()
- Three Key Structural Technologies
X-Ray, MicroED and CryoEM-SPA solve all structural biology challenges.
-
![]()
- One-Stop Service
Comprehensive gene-structure biology services, addressing all structural biology needs.
Service Details
|
Steps
|
Service Details
|
Turnaround Time
|
|
AI Design & Optimization
|
• AI Sequence Design
• Sequence Analysis
• Codon Optimization
• Screening Proteins
|
3-5 Days
|
|
Gene Synthesis
|
• Gene Synthesis
• Vector Construction
|
Start from 1 Week
|
|
Protein Expression & Purification
|
• Protein Expression Validation
• Large-scale Cultivation
• Protein Purification
|
Start from 2 Weeks
|
|
Protein Structural Analysis
|
• Protein Crystallization Optimization
• X-Ray Diffraction
• MicroED
• CryoEM-SPA
|
Start from 1 Month
|
|
Large-Scale Production
|
• Provide Bacteria for Large-scale Fermentation
• Provide Expressed Proteins up to Gram Level
|
Quote
|
Deliverables
-
2~5 µg lyophilized plasmid DNA
-
Sequencing chromatogram
-
1- 5 mg protein products
-
COA report
-
The report, structure factor data, and atomic coordinates conforming to PDB submission requirements.
Case Study
X Ray Diffraction
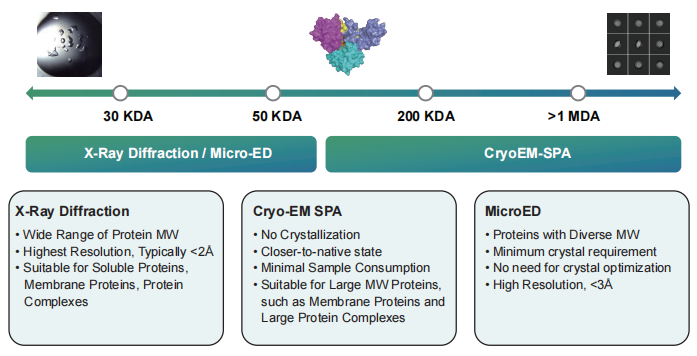
▷ KRAS G12D
Technical difficulty:
-
KRAS binds to GTP and is in an activated state; when it binds to GDP, it switches to an off-state. The conversion of GTP to GDP is an extremely rapid process, making it difficult to obtain the activated state of KRAS bound to GTP.
-
To study this target, it is necessary to obtain the protein in both the activated and off states.
Solution:
-
We degraded the GDP bound to KRAS using an enzyme, then added GppCp and incubated it with the KRAS protein to obtain the activated state of the KRAS protein. Finally, we solved the high-resolution structures of KRAS G12D-GDP/GppCp and KRAS G12D-GDP-1133 (~1.8 Å).
MicroED
▷ IL5RA
Technical difficulty:
-
IL5RA is highly prone to forming inclusion bodies during protein expression, typically appearing in an insoluble form.
-
The cocrystals of IL5RA and peptides are extremely small (<10 μm), making it very difficult to optimize and obtain larger single crystals (>50 μm).
Solution:
-
We purified IL5RA inclusion bodies and refolded the protein to obtain soluble IL5RA with >90% purity.
-
We mounted the crystals on copper grids, prepared 200 nm-thick sections using Cryo-FIB, and collected diffraction data via MicroED. The structure was solved at 3 Å resolution, with clear electron density for the AF17121 peptide.

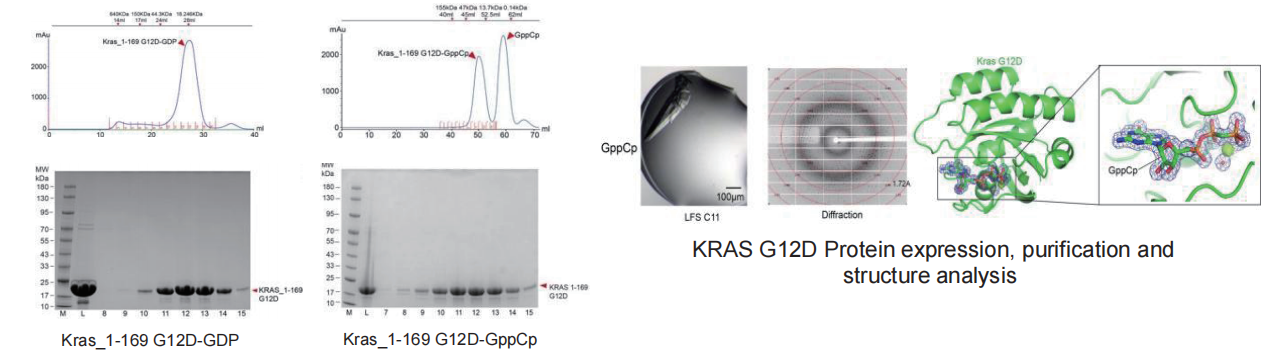
Cryo-EM SPA
▷ GPCR – GCGR
Technical difficulty:
-
The transmembrane regions of membrane proteins possess extensive hydrophobic surfaces, making them difficult to stabilize in polar aqueous solutions after dissociation from the membrane.
-
The protein expression level of GCGR is extremely low, which prevents the completion of subsequent purification.
-
In the protein sample preparation stage, the protein samples mostly adhere to the supporting film on the grid rather than being embedded in the ice layer within the holes of the supporting film.
-
The protein samples exhibit significant preferential orientation.
Solution:
-
We changed the expression vector and protein expression system to increase protein yield; and added monoclonal fragments during expression to enhance the stability of the complex.
-
During the protein sample loading process, we adjusted the protein concentration and tried other types of grids, eventually achieving a higher number of protein particles embedded in the holes.
-
Increased the thickness of the vitreous ice layer and applied a carbon film on the grid to average the number of particles in different orientations, thereby improving the preferential orientation of the protein particles.
FAQs

Analyzing a protein’s structure involves examining its shape and folding across multiple levels using both experimental and computational tools. The process usually begins by purifying the protein sample, followed by techniques to reveal how the atoms are arranged.
-
Laboratory methods like X-ray crystallography and cryo-electron microscopy (cryo-EM) are used to capture detailed 3D structures. X-ray crystallography requires the protein to form crystals, while cryo-EM freezes proteins in solution for imaging.
-
Computational approaches, especially AI-based prediction models like AlphaFold, can infer likely 3D shapes from amino acid sequences.
-
Spectroscopic tools such as circular dichroism (CD) help identify the presence of specific secondary structures like helices or sheets.
Together, these approaches provide insights into how the protein folds, how stable it is, and how it may interact with other molecules.
There are several methods tailored to understanding different levels of protein structure:
-
Primary structure (amino acid sequence): Identified using mass spectrometry or sequencing.
-
Secondary structure: CD spectroscopy and Fourier-transform infrared spectroscopy (FTIR) help detect common structural motifs like alpha-helices and beta-sheets.
-
Tertiary and quaternary structures:
⊙ X-ray crystallography: Offers atomic resolution but requires protein crystallization.
⊙ Nuclear magnetic resonance (NMR): Works in solution but is best for smaller proteins.
-
Cryo-EM: Ideal for large complexes and doesn’t require crystals.
-
Cross-linking and mass spectrometry: Reveal spatial relationships between different parts of a protein.
-
In silico modeling: Tools like homology modeling and machine learning-based predictions fill gaps where experimental data is lacking.
Often, a combination of these techniques provides the most complete picture.
Protein structure is categorized into four hierarchical levels, each identifiable using different methods:
-
Primary structure (linear sequence of amino acids): Determined through sequencing or mass spectrometry.
-
Secondary structure (local folding patterns): CD or infrared spectroscopy can estimate proportions of helices, sheets, and turns.
-
Tertiary structure (3D folding of a single polypeptide): Solved through high-resolution tools like crystallography, NMR, or cryo-EM, or predicted computationally.
-
Quaternary structure (multi-subunit assemblies): Studied via techniques that reveal subunit interactions, such as size exclusion chromatography, light scattering, or cryo-EM.
A combination of these strategies gives a full understanding of how a protein is organized from its sequence to its functional architecture.
Get in Touch with Us
Related Services
 DNA Synthesis
DNA Synthesis Vector Selection
Vector Selection Molecular Biology
Molecular Biology Oligo Synthesis
Oligo Synthesis RNA Synthesis
RNA Synthesis Variant Libraries
Variant Libraries Genome KO Library
Genome KO Library Oligo Pools
Oligo Pools Virus Packaging
Virus Packaging Gene Editing
Gene Editing Protein Expression
Protein Expression Antibody Services
Antibody Services Peptide Services
Peptide Services DNA Data Storage
DNA Data Storage Standard Oligo
Standard Oligo Standard Genome KO Libraries
Standard Genome KO Libraries Standard Genome Editing Plasmid
Standard Genome Editing Plasmid ProXpress
ProXpress Protein Products
Protein Products