DNA Synthesis
DNA Synthesis Vector Selection
Vector Selection Molecular Biology
Molecular Biology Oligo Synthesis
Oligo Synthesis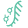 RNA Synthesis
RNA Synthesis Variant Libraries
Variant Libraries Genome KO Library
Genome KO Library Oligo Pools
Oligo Pools Virus Packaging
Virus Packaging Gene Editing
Gene Editing Protein Expression
Protein Expression Antibody Services
Antibody Services Peptide Services
Peptide Services DNA Data Storage
DNA Data Storage Standard Oligo
Standard Oligo Standard Genome KO Libraries
Standard Genome KO Libraries Standard Genome Editing Plasmid
Standard Genome Editing Plasmid ProXpress
ProXpress Protein Products
Protein Products
In the exciting frontier of biotechnology, a revolutionary force is at work - Artificial Intelligence (AI) in the realm of proteins. AI is revolutionizing protein research. It enables scientists to predict protein structures and functions with unprecedented accuracy, design novel proteins from scratch, and unlock the molecular mysteries of life. The potential of this technology is limitless, paving the way for new possibilities in drug discovery, bioengineering, and beyond. Advancements in protein production, such as AlphaFold and AI-driven technologies, have not only rewritten the rules of protein research - but have also opened many possibilities in various fields from drug development to disease diagnosis and synthetic biology.
AI’s Role in Protein Structure Prediction
Protein research has long been a core challenge in the field of biotechnology, primarily due to the diverse three-dimensional structures and dynamic properties of proteins. For decades, scientists have been using traditional tools like X-ray crystallography, nuclear magnetic resonance (NMR), andCryo-electron microscopy to decipher protein structures. However, these methods often prove to be costly, time-consuming, and sometimes ineffective - especially when dealing with new and complex proteins. These traditional techniques also face significant challenges in addressing the dynamic and environment-dependent nature of proteins, often resulting in structural analyses that may not accurately represent the state of proteins under physiological conditions. There is an urgent need for new tools that can predict and resolve complex protein structures more quickly, efficiently, and cost-effectively to meet the demands of modern life science research.
How to break through?
As computer technology and algorithms continue to advance, scientists have begun to explore the use of artificial intelligence and machine learning methods to solve the problem of protein structure prediction.
In 2020, DeepMind introduced the groundbreaking tool called AlphaFold, which has since undergone continuous development and improvement. This model leverages deep learning to accurately predict the three-dimensional structures of proteins, significantly enhancing both the accuracy and speed of protein structure prediction while bringing revolutionary progress to biological research. AlphaFold uses a deep learning model to analyze evolutionary covariation data from multiple sequence alignments (MSA), predicting distances between amino acid residues and thereby revealing the spatial configuration of protein structures. The neural network predicts a “distogram,” or a probabilistic map of residue-residue distances, which guides the folding process. Using this distance information, the model applies optimization (such as gradient descent) to determine the final 3D structure of the protein.
In 2020, AlphaFold 2.0 made a remarkable debut at the CASP14 protein structure prediction competition, achieving an unprecedented 98.5% accuracy in predicting protein structures. As an enhanced version of the original AlphaFold, this breakthrough model can predict the structure of a typical protein in just minutes, with an accuracy often within 1 Å—about the width of a carbon atom (roughly 1.4 Å). This level of precision represents a significant leap forward in computational biology, offering a powerful tool for understanding the complexities of protein folding.
AlphaFold 2.0 introduced an advanced neural network architecture called Evoformer. This architecture processes multiple sequence alignments (MSAs) and pairwise residue information, leveraging evolutionary and spatial relationships. With triangle updates and attention mechanisms, Evoformer enables the model to capture long-range dependencies and spatial relationships, critical for accurate protein folding predictions. It also includes a structure module that directly predicts 3D atomic coordinates and employs a recycling mechanism for iterative refinement. By using end-to-end training and a unique self-distillation process, AlphaFold 2.0 achieves near-experimental accuracy, allowing it to predict complex and novel structures that were previously challenging.
In 2021, David Baker’s team introduced the revolutionary tool RoseTTAFold. This open-source protein structure prediction tool features a unique three-track network architecture, enabling it to simultaneously process sequence, distance, and coordinate information - which significantly enhances prediction accuracy and speed.
-
Sequence Track: Handles amino acid sequence information.
-
Distance Track: Handles information on interactions between amino acid pairs.
-
Coordinate Track: Handles local structural features, such as secondary structure and solvent accessibility.
This model simultaneously processes sequence, distance, and coordinate information in separate channels, allowing iterative refinement of protein structures through continuous exchange of information between channels. RoseTTAFold’s design enables a multi-task learning approach to simultaneously optimize multiple related tasks, like distance map prediction, angle map prediction, and contact map prediction. This approach helps to improve the overall prediction accuracy.
|
Year |
Model |
Team |
Architecture |
Applications |
Limitations |
|---|---|---|---|---|---|
|
2018 |
AlphaFold |
DeepMind |
Input: Multiple sequence comparison information Algorithm: Using deep neural networks, predict the properties of amino acid chains, such as the distribution of distances between two and two, the distribution of angles, etc. |
Protein Structure Prediction |
Insufficient prediction accuracy to predict orphan protein structures |
|
2020 |
AlphaFold2 |
DeepMind |
Input: Multiple sequence comparison information Algorithm: A transformer architecture using attention to learn features from the network to predict 3D structure coordinates |
Protein Structure Prediction |
Weak predictive power in recognizing protein-aptamer and interactions |
|
2021 |
RoseTTAFold |
University of Washington, David Baker Team |
Input: multiple sequence comparison information Algorithm: use NLP method to learn co-evolutionary information directly from MSA to build protein 3D structure model |
Protein Structure Prediction, Protein-Protein Complexes |
Smaller model size compared to AlphaFold2, lack of sidechain coordinate inference |
AI’s Role inProtein Function Prediction
Protein functions are defined within the Gene Ontology (GO), which organizes proteins by their molecular functions (MFO), roles in biological processes (BPO), and locations in cellular components (CCO). Through annotations that often extend to homologous proteins, resources like UniProtKB/Swiss-Prot provide curated GO data for thousands of organisms and over 550,000 proteins.
Yet, most proteins in the database lack functional annotations, with existing annotations derived mainly from time-consuming experiments. AI-based prediction methods, integrating amino acid sequences, structural information, and protein-protein interaction (PPI) networks, offer a scalable solution to bridge this gap. By leveraging deep learning and literature insights, these tools enable more efficient and precise functional predictions, deepening our understanding of protein roles in health and disease.
DeepGO is the first deep learning-based predictive model that predicts protein functions by applying deep learning to protein sequence and interaction data. The model takes as input the amino acid sequences of proteins, from which it extracts features using convolutional neural networks (CNNs). DeepGO also incorporates protein-protein interaction (PPI) networks, which allow the model to leverage functional relationships between proteins. Through this approach, DeepGO assigns Gene Ontology (GO) terms that categorize protein functions based on molecular function, biological process, and cellular component - making it useful for comprehensive protein function prediction.
DeepGO-SE is an advanced model for protein function prediction that incorporates knowledge from Gene Ontology (GO) through a process called approximate semantic entailment. The model operates in three key steps:
1. Constructing Approximate Models: Using GO’s axioms and protein function assertions, DeepGO-SE creates approximate models, where ELEmbeddings capture the semantic relationships within GO as geometric representations.
2. Protein Embedding and Optimization: Protein sequences are represented with embeddings from the pretrained ESM2 model. These embeddings are then positioned within the approximate models to maximize the likelihood of statements like “protein has function C,” guiding precise function predictions.
3. Multi-Model Aggregation: This process is repeated to generate multiple models, and the final prediction is based on truth values that hold across all models, effectively capturing the entailment.
|
Year |
Model |
Journal |
Architecture |
Limitations |
|---|---|---|---|---|
|
2018 |
DeepGO |
Bioinformatics |
Input: Protein sequence and PPI network; |
Hierarchical classification networks require huge memory resources and are difficult to apply to large-scale labeling. |
|
2024 |
DeepGO-SE |
Nature Machine Intelligence |
Input: Protein sequences Algorithm: Prediction of GO functions from protein sequences using a pre-trained large language model, by generating multiple approximate GO models and predicting the true value of the protein function using a neural network. |
Best performance when sequences bind to PPIs, many proteins have no known interactions limiting the application of combinatorial models. |
AI’s Role in Protein Design
Protein design has evolved significantly, moving from PCR-based mutagenesis, where researchers introduced specific mutations to alter protein structure, to advanced computational methods capable of constructing novel proteins with desired features. Today, with rapid advances in structural biology, computational modeling, and AI, protein design is more precise and accessible than ever. AI-powered design technologies now enable bothprotein optimization, enhancing natural protein functions like affinity and stability, and de novo design, creating entirely new proteins with tailored functions, structures, and applications.
ProteinMPNN is a deep learning model for efficient protein sequence design, bypassing the computational demands of traditional physics-based methods like Rosetta. By learning directly from structural data and encoding spatial relationships, ProteinMPNN accurately predicts sequences that fold into target structures. The model has shown strong performance in designing complex assemblies, such as tetrahedral nanoparticles, and in maintaining binding affinity even with point mutations. With its ability to create diverse protein types, including monomers, assemblies, and nanoparticles, ProteinMPNN is a powerful tool for enhancing protein solubility, stability, and functionality.
RFdiffusion: RFdiffusion takes an innovative approach using diffusion models to refine protein backbones from initial noise to realistic structures through iterative denoising. By incorporating specific structural motifs, it creates diverse, complex proteins tailored to specific needs, such as symmetric assemblies and functional motifs. RFdiffusion’s precision in generating novel topologies enables cutting-edge applications, from therapeutic scaffolds to complex structural designs.
ProGen: Adapting language model techniques from NLP, ProGen generates protein sequences that reflect evolutionary patterns and biochemical properties, enabling function-specific sequence creation. ProGen uses transformers to model sequence features that drive stability and functionality, making it highly adaptable for various applications, from enzyme design to generating sequences with specific binding affinities.
AlphaProteo: Focused on creating high-affinity protein binders, AlphaProteo uses structure-guided sequence generation to develop binders targeting precise protein sites. By combining generative modeling with advanced filters, AlphaProteo excels in producing binders for challenging targets like viral and cancer-related proteins, with sub-nanomolar affinities achieved in many cases. Its ability to streamline binder design with minimal rounds of optimization opens new possibilities in therapeutic development.
AlphaProteo has a higher experimental success rate on the seven target proteins tested. In wet-lab testing, 9% to 88% of candidate molecules successfully bound, which is 5 to 100 times higher than other methods; and 3 to 300 times higher than the binding affinity of the best available method.
From AI Design to Wet Lab Validation
Researchers now have the ability to generate proteins optimized for specific functions like binding affinity, stability, and catalytic activity directly from sequence and structural data. However, transforming these computational designs into functional, reliable proteins requires rigorous wet-lab validation to ensure properties such as binding affinity, stability, and bioactivity. During the wet lab validation process, scientists encounter numerous challenges. The complex structures and properties of certain proteins can lead to misfolding or the formation of inactive aggregates during expression, significantly reducing expression efficiency. Data feedback from wet lab experiments is crucial, as it can be used to further optimize AI models, enhancing their accuracy and effectiveness in future protein designs.
Synbio Technologies offers a complete one-stop solution for researchers, simplifying the journey from digital sequence to experimentally validated protein products. With our services, you only need to provide the protein sequence - Synbio handles every detail, from codon optimization and gene synthesis to expression system selection, protein purification, and functional validation.
Our one-stop solution includes:
-
Codon Optimization: Enhance expression with our NG Codon technology, optimized for your specific expression system.
-
Gene Synthesis & Cloning: High-fidelity gene synthesis and cloning into any specified vector.
-
Expression System Screening: Access to bacterial, yeast, insect, and mammalian hosts for tailored expression.
-
Recombinant Antibody Expression: Cover all stages of antibody discovery by integrating antibody genes sequence,de novo antibody design, antibody humanization, antibody genes synthesis, recombinant antibody expression, monoclonal antibody preparation and polyclonal antibody preparation. .
-
Scalable Protein Production: Flexible production options, from micrograms to grams, to support projects of any size.
-
Quality & Functional Validation: Comprehensive testing ensures reliability, activity, and functionality of the final protein or antibody product.
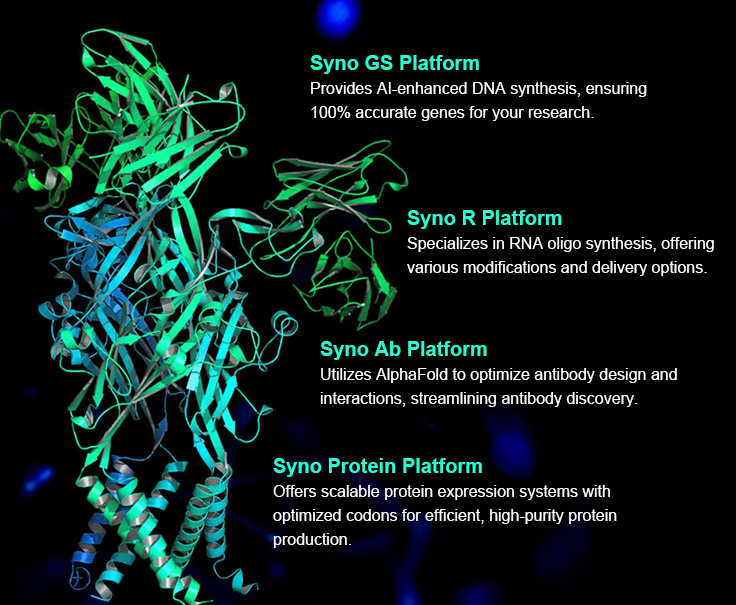
Global AI Protein Companies
Partial companies, Contact Us with the message“AI Protein Company List”for more information to download the full version
The versatility of AI applications in protein science is already reshaping drug discovery, precision medicine, and synthetic biology. Companies incubated by pioneers like David Baker are using deep learning to address key biological challenges, including drug development, peptide design, small-molecule binding protein engineering, and novel material synthesis. With AI’s proven ability to accelerate and enhance protein design, its role in biotech is expected to grow, unlocking new possibilities for tailored therapies and innovative biomaterials, and pushing the boundaries of what’s possible in the life sciences.
References
[1] Jumper, John, et al. "Highly accurate protein structure prediction with AlphaFold." nature 596.7873 (2021): 583-589.
[2] Baek, Minkyung, et al. "Accurate prediction of protein structures and interactions using a three-track neural network." Science 373.6557 (2021): 871-876.
[3] Madani, Ali, et al. "Large language models generate functional protein sequences across diverse families." Nature Biotechnology 41.8 (2023): 1099-1106.
[4] Senior, Andrew W., et al. "Improved protein structure prediction using potentials from deep learning." Nature 577.7792 (2020): 706-710.
[5] Zambaldi, Vinicius, et al. "De novo design of high-affinity protein binders with AlphaProteo." arXiv preprint arXiv:2409.08022 (2024).
[6] Kulmanov, Maxat, et al. "Protein function prediction as approximate semantic entailment." Nature Machine Intelligence 6.2 (2024): 220-228.
[7] Watson, Joseph L., et al. "De novo design of protein structure and function with RFdiffusion." Nature 620.7976 (2023): 1089-1100.